We're thrilled to announce that LazySlide has been published in Nature Methods! This milestone marks the culmination of years of development, community engagement, and a vision to make computational pathology accessible to everyone—from single-cell genomics researchers to clinical pathologists.
![]()
In this post, we want to take you behind the scenes: the motivation that drove us, the technical decisions we made, and the exciting applications that LazySlide now enables. We hope this provides insight into our development philosophy and inspires others working at the intersection of AI and biology.
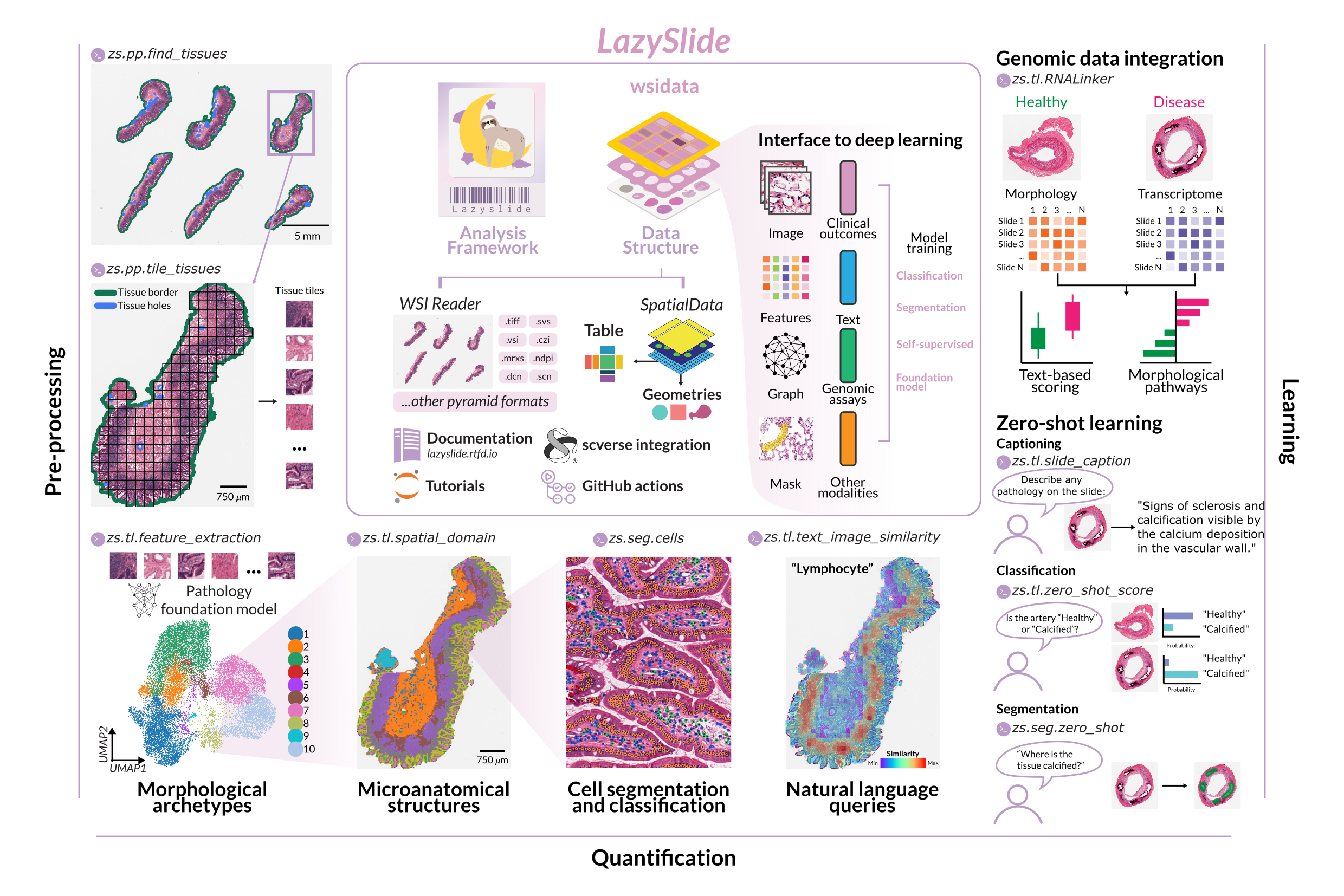
The problem we set out to solve
A revolution in vision AI
The past few years have witnessed a remarkable transformation in computer vision. Self-supervised learning has enabled the training of powerful visual encoders on massive unlabeled datasets. In histopathology, this has led to an explosion of foundation models—CONCH, UNI/UNI2, Virchow/Virchow2, Prov-GigaPath, PRISM, TITAN, and dozens more—each promising to extract rich, biologically meaningful representations from tissue images.
Yet this abundance created its own problem: fragmentation. Each model came with its own preprocessing requirements, input formats, and inference pipelines. Researchers wanting to compare models or integrate them into existing workflows faced a daunting task of writing custom glue code for each one.
The single-cell community 'looks at tissue'
Meanwhile, the genomics and single-cell community was increasingly interested in spatial biology. Spatial transcriptomics technologies were maturing rapidly, and researchers were asking deeper questions about how cells organize within tissues. But there was a curious gap: while spatial transcriptomics captured molecular profiles at specific locations, histopathology—the oldest and most widely available source of spatial tissue data—remained largely inaccessible to this community.
Why? Histopathology analysis required specialized tools, different data structures, and expertise that most genomics researchers didn't have. The irony was stark: billions of H&E-stained slides exist in pathology archives worldwide, representing an unparalleled resource for understanding tissue biology at scale. Yet the communities best equipped to analyze high-dimensional biological data were effectively locked out.
Our vision
We asked ourselves: What if analyzing a whole slide image was as straightforward as analyzing a single-cell dataset?
This meant building a framework that:
- Spoke the language of the genomics community (AnnData, scverse tools)
- Abstracted away the complexity of foundation models
- Scaled from a single slide to thousands
- Enabled both novices and experts to work productively
LazySlide was born from this vision.
Building LazySlide: technical philosophy
Modern Python, modern tooling
From the start, we committed to modern Python practices. We use uv for fast, reproducible package management. Our CI/CD pipeline runs comprehensive tests on every commit. Type hints throughout the codebase catch errors early and improve IDE support.
This isn't just about developer ergonomics—it's about sustainability. Foundation models evolve rapidly. New architectures appear monthly. A well-structured codebase lets us incorporate new models quickly without breaking existing functionality.
Separation of concerns
LazySlide is actually part of a broader ecosystem we've built:
- WSIData: The core data structure, extending AnnData for whole slide images
- LazySlide: The analysis framework with all operations and model integrations
- LazySlide-tutorials: Comprehensive learning resources
- LazySlide-benchmark: Reproducible performance comparisons
This separation means researchers can use just what they need. Want only the data structure for your own pipeline? Use WSIData. Need to run at scale on a cluster? The Nextflow pipeline has you covered.
44 foundation models and counting
Perhaps our most ambitious undertaking: LazySlide now supports 44 foundation models across classification, segmentation, and multimodal tasks. You can see the full list in our documentation.
Each model required understanding its specific requirements—input normalization, patch sizes, embedding dimensions—and wrapping them in a consistent interface. The result is that switching between models is often a single parameter change:
## Extract features with the UNI model
zs.tl.extract_features(wsi, model="uni")
## Or try CONCH instead
zs.tl.extract_features(wsi, model="conch")
This uniformity enables systematic comparisons that were previously impractical.
A note on foundation models and licensing
LazySlide's power comes from integrating dozens of foundation models developed by research groups worldwide. We are deeply grateful to the authors of these models for making their work available to the community.
Important: The usage of any model in LazySlide is subject to the terms and conditions of the respective model's license. Please ensure you comply with the license terms before using any model. If you use a model in your research, please cite the original paper or repository as appropriate.
LazySlide does not redistribute any source code that's not compatible with LazySlide's MIT license. We simply provide a unified interface to access these models—the responsibility for proper usage, citation, and license compliance rests with the user.
You can find license information for each model in our model zoo documentation.
Applications: what's now possible
LazySlide doesn't just make existing workflows easier—it enables entirely new analyses that bridge computational pathology with genomics and natural language understanding.
Simplicity first
Before diving into advanced capabilities, let's start with the basics. LazySlide was designed so that a complete preprocessing pipeline—tissue segmentation, tessellation, and feature extraction—can be run in just a few lines of code:
import lazyslide as zs
wsi = zs.datasets.sample()
# Pipeline
zs.pp.find_tissues(wsi)
zs.pp.tile_tissues(wsi, tile_px=256, mpp=0.5)
zs.tl.feature_extraction(wsi, model='resnet50')
# Access the features
features = wsi['resnet50_tiles']
# Visualize the 1st and 99th features
zs.pl.tiles(wsi, feature_key="resnet50", color=["1", "99"])
This entire workflow runs in approximately 7 seconds on a MacBook Pro. No complex configuration, no boilerplate code—just intuitive, scanpy-style commands that feel familiar to anyone who has worked with single-cell data.
This simplicity isn't accidental. Our benchmarking showed that LazySlide completes standard preprocessing workflows with fewer lines of code, lower token count, and a simpler API compared to established tools, facilitating rapid development and code maintenance [1]. The framework supports essential tasks such as tissue segmentation, cell classification, and morphological archetype analysis with minimal setup—essentially one command each—empowering researchers with little experience in digital pathology [2].
But simplicity doesn't mean limited capability. LazySlide scales from quick exploratory analyses to production pipelines processing thousands of slides. Let's look at some of the more advanced applications this foundation enables.
Zero-shot learning
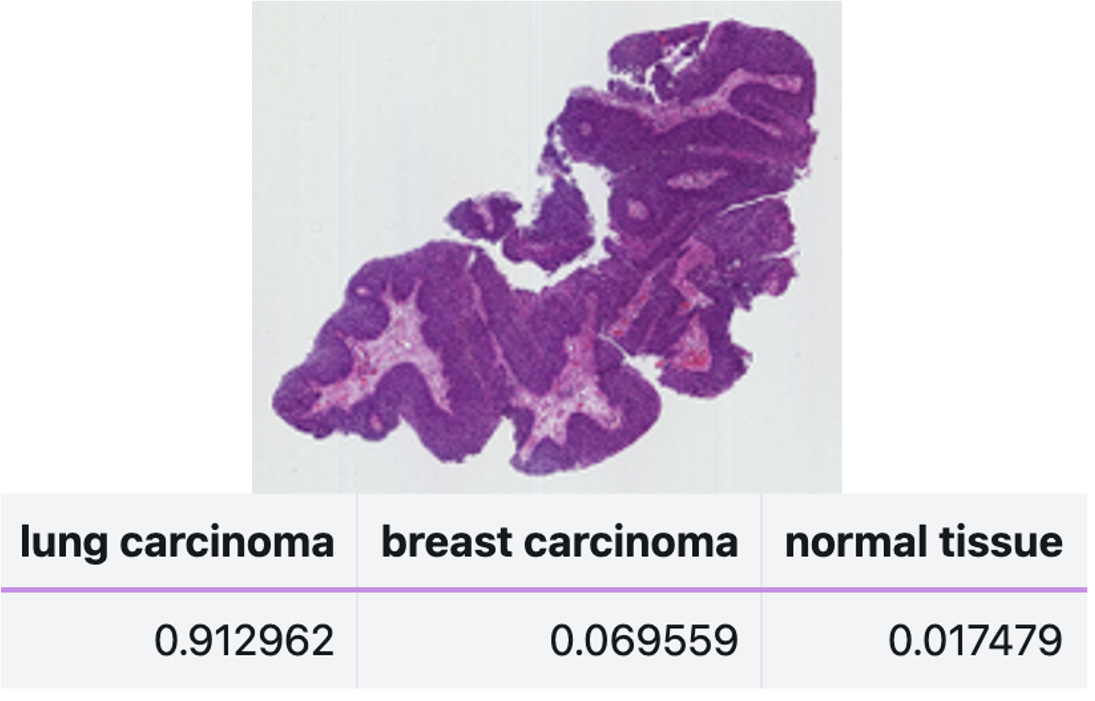
Traditional machine learning requires labeled training data. But what if you could classify tissue based on natural language descriptions alone?
LazySlide supports zero-shot learning through multimodal vision-language foundation models such as PRISM and TITAN [1]. You can compare slide embeddings to arbitrary text prompts:
## Classify without any training data
probabilities = zs.tl.zero_shot_score(
wsi,
prompts=["lung carcinoma", "breast carcinoma", "normal tissue"],
feature_key="virchow",
)
This is transformative for rare diseases or novel phenotypes where labeled data simply doesn't exist.
Text-guided semantic segmentation
Taking zero-shot further, LazySlide enables segmentation guided by natural language. Want to find all regions matching "dense inflammatory infiltrate"? LazySlide uses text-image similarity metrics to produce binary masks, then refines them with SAM2 for precise boundaries.
## Segment tissue based on text description
mask = zs.seg.zero_shot(
wsi,
prompt="dense inflammatory infiltrate",
table_key="conch_tiles_text_similarities",
threshold=0.5
)
This bridges the gap between how pathologists describe tissue and how computers analyze it.
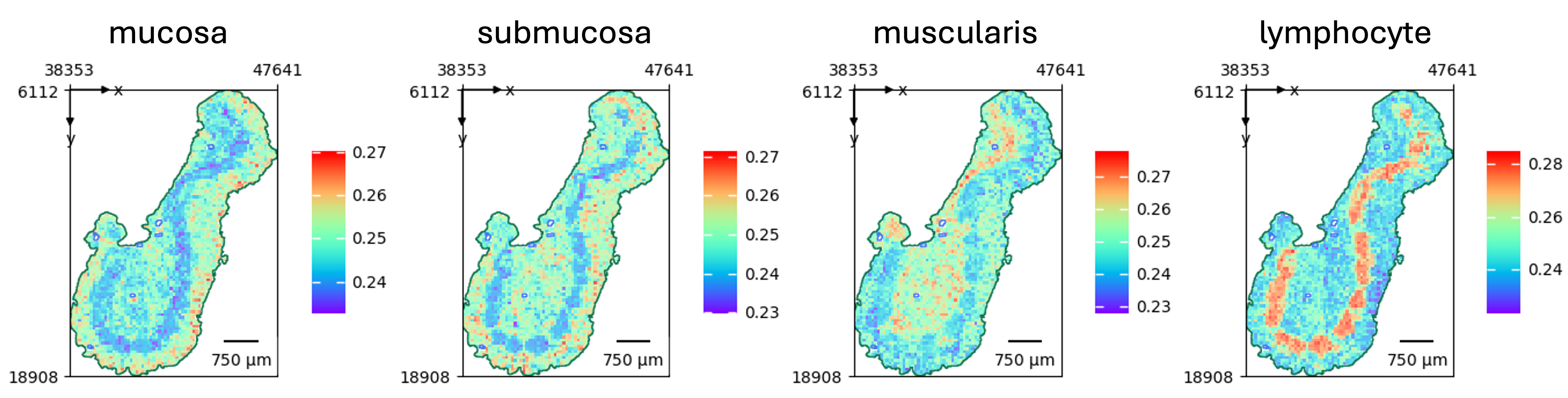
Natural language queries
One of the most powerful features leverages multimodal foundation models to enable content retrieval through text-based queries [1]. The framework employs pathology-specific models such as PLIP and CONCH to facilitate semantic search within whole slide images [2]. Users can search for specific histological patterns, cell types, or tissue structures using natural language descriptions.
For example, in our analysis of human artery slides from the GTEx project, we demonstrated how terms related to "calcification" show higher enrichment in calcified samples, whereas anatomical terms predominate in healthy tissues [1]. A differential analysis highlighted terms such as gap junction, vascular niche, and apoptosis as significantly enriched in calcified arteries—consistent with observed morphological changes [1].
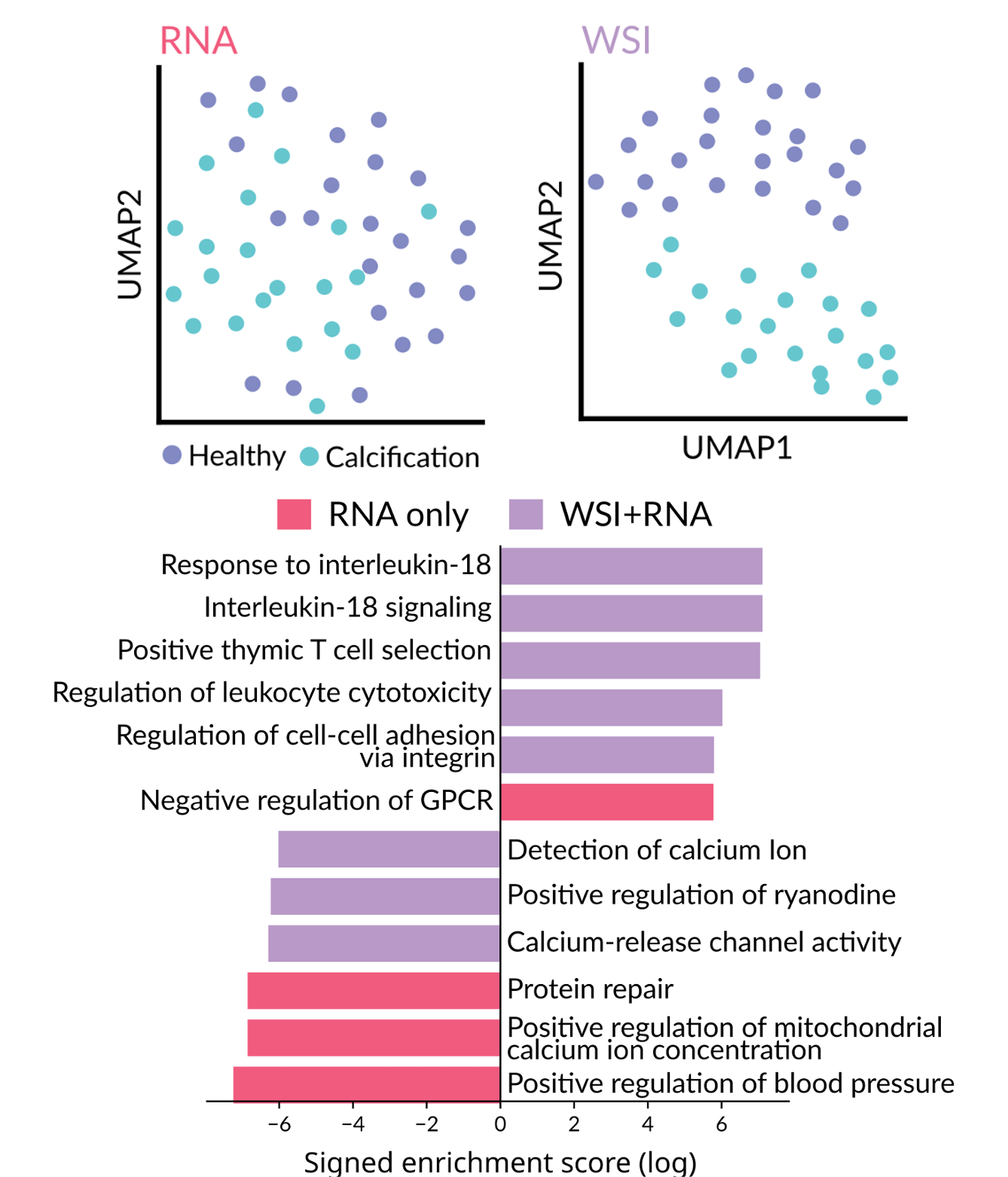
Integration with bulk RNA-seq and other genomic modalities
One of our most exciting applications connects histology with transcriptomics. Given paired H&E images and bulk RNA-seq data, LazySlide can learn relationships between morphological features and gene expression to deliver more insight than RNA-seq alone.
Moreover, LazySlide can predict protein expressions from HE images. This opens possibilities for mining the vast archives of H&E slides that lack molecular profiling, imputing molecular information from morphology [2].
Natural language summaries
For descriptive analysis, LazySlide can generate natural language summaries of histological content from slide embeddings. Imagine processing thousands of slides and getting preliminary text reports automatically—not to replace pathologists, but to assist them with concise, context-aware interpretations.
Benchmarking: how does it compare?
We believe in rigorous, reproducible benchmarking. Our dedicated benchmark repository compares LazySlide against six established frameworks and QuPath, the gold standard in pathology analysis.
The results show that LazySlide achieves comparable or superior classification accuracy while dramatically reducing code complexity. Tasks that require dozens of lines in other frameworks often need just one or two in LazySlide.
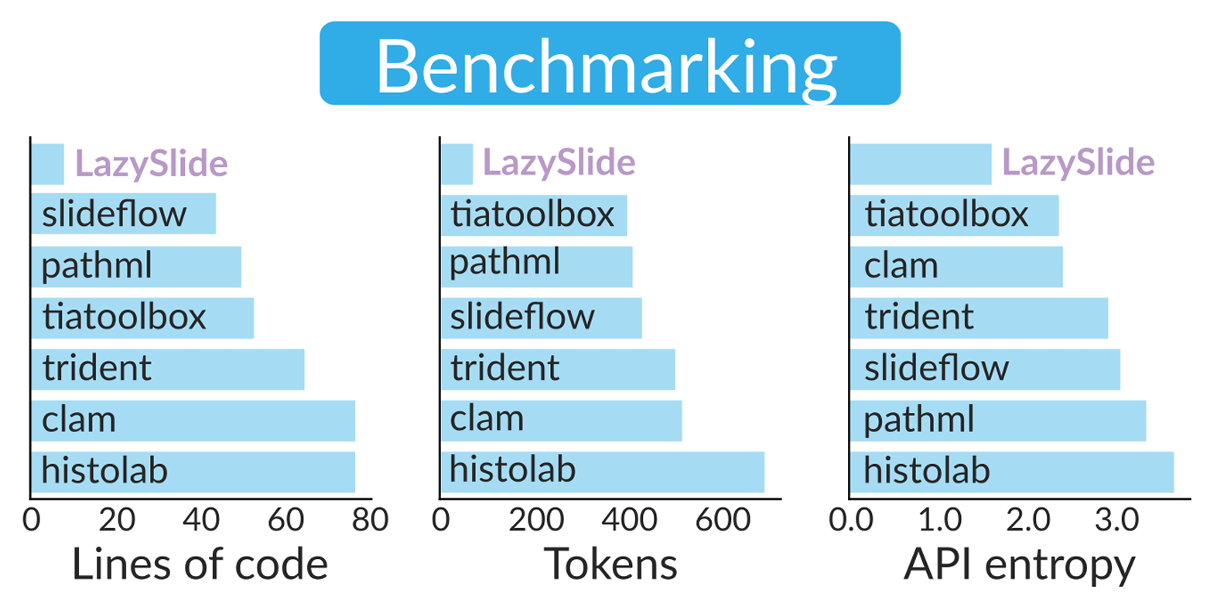
Community and adoption
Part of scverse
LazySlide is officially part of the scverse ecosystem, the community-driven collection of tools for single-cell and spatial omics. This means seamless integration with scanpy, squidpy, and the broader Python biology stack.
Given its maturity, open-source availability, and design philosophy focused on modularity and ease of use, LazySlide fills a critical gap by making digital pathology accessible to researchers experienced in single-cell analysis who seek to expand their toolbox with histological methods powered by foundation models [2].
Spreading the word
Yimin has been tireless in presenting LazySlide to the community:
- scverse conference: community showcase of LazySlide (yes, at late-late-night hours!)
- scverse community meeting: invited talk
- Sydney Statistical Bioinformatics Seminar: "The lazy way of pathological image analysis"
- BioLinkX Workshop at Zhejiang University: invited talk
- European Society of Pathology meeting: e-poster presentation
- Cell Symposia on Hallmarks of Cancer: poster presentation
And yes, we made stickers. Very cool stickers. The sloth mascot has been a hit.

Rapid adoption
The community response has been overwhelming. You can see the growth trajectory on Star History:
We hit 100 GitHub stars in July 2025, and growth has only accelerated since. More importantly, we're seeing LazySlide cited in preprints, used in workshops, usage via MCP servers, and integrated into research pipelines worldwide.
What's next
Publication in Nature Methods is not an endpoint—it's a milestone. We're actively working on:
- More foundation models: the field moves fast, and we're committed to keeping up
- Enhanced multimodal capabilities: deeper integration between vision, language, and molecular data
- Clinical deployment tools: making LazySlide ready for regulated environments
- Community contributions: we welcome PRs and are building contributor documentation
Try it yourself
Ready to get started?
pip install lazyslide
Then explore the resources:
- 📚 Documentation
- 🎓 Tutorials
- 💻 Source code
- 📊 Benchmarks
Acknowledgments
LazySlide exists because of the incredible work of the entire team, with Yimin Zheng leading the development effort. We thank Elisabeth Weigert, Elisabeth Gurnhofer, Patrick Wagner, and Gerald Timelthaler for their technical support and pathologists Zsuzsanna Bagó-Horváth and Ulrike Heber for their feedback on tissue annotations [1].
We're grateful to the scverse community for their support and for welcoming LazySlide into the ecosystem [2]. We also owe a debt of gratitude to the developers of the 44 foundation models we integrate—their open science practices make frameworks like LazySlide possible.
Finally, thank you to everyone who has tested LazySlide, reported bugs, suggested features, and spread the word. Building open-source scientific software is a community endeavor. Thank you for being part of it.
LazySlide is open source under the MIT license. If you use it in your research, please cite our Nature Methods paper.